作者:刘芷若,冯博,中国医科大学附属第一医院放射科
病例女,70岁,于1年前偶然发现背部肿物,无疼痛,未经治疗,肿物逐渐增大,近2月邻近背部肿物处又发现一肿物。专科检查发现背部脊柱两侧约平第12胸椎及第3腰椎可见局限性隆起,皮肤不红,可触及范围约4cm×3cm大小肿物,边界清,质地韧,活动差。病来自觉活动后气短。超声:皮下不均质回声包块。
手术及病理:背部肿物局部切除送病理,灰红色组织,结节状,部分包膜,局部钙化,切面灰白色,质韧。免疫组化:CD56(-),SMA(-),Desmin(-),MyD1(-),S-100(-),CD34(-),TLE1(-),TFE3(-),CK7(-),CD99(-),PAX-5(-),EMA(-),网状纤维(-),CK广谱(-),Vimentin(+),Ki-6730%(+),INI1(+),Mum1(+),CD3(+),CD20(+),CD138(+),CD38(+);骨髓

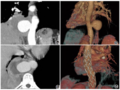
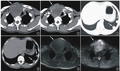
图1前纵隔软组织密度团块影,边界不清。图2,3左侧后胸壁、胸椎旁软组织内亦可见不规则团块影,左侧胸腔少量积液。图4背部皮下脂肪层内见类似密度影,边界清。图5镜下可见瘤细胞较丰富,多角形为主,无明显异型性,偶见核分裂像。图6化疗两个月后复查CT病变数量减少,范围明显缩小。
讨论
浆细胞瘤是起源于B淋巴细胞、以浆细胞单克隆增殖为特征的的一组疾病,包括
EMP好发于男性,可发生在各个年龄段,平均发病年龄60岁。由于浆细胞在全身广泛分布,任何有淋巴网状组织的部位均可发生EMP。好发部位为头颈部,尤其是上呼吸道黏膜(如鼻腔、鼻窦及鼻咽部),口腔、消化道、椎旁软组织、肺部、纵隔、肾脏等亦可发生。部分EMP病例后期可转化为MM,且许多病例往往在发展为MM时才发现从而导致误诊。
组织学上,SPB组织由分化程度各异的肿瘤性浆细胞组成,根据后者分化程度可将SPB分为低、中、高度恶性三级,本例病例为偶见核分裂像的Ⅰ级(低度恶性)。免疫组织化学方面髓外浆细胞瘤通常CD79α、CD38、CD138、Lambda/Kappa阳性表达,Ki-67增殖指数约40%,CD3、CD5和CD20常为阴性,本例病理与文献描述基本相符。
由于EMP通常以不同部位的软组织肿块为首发症状,临床上表现为疼痛或邻近组织受压症状,从而极易被误诊为其他性质的占位性病变。目前国内外文献均普遍认为EMP临床和影像学表现缺乏特征性,并且EMP实验室检查结果价值不大,血液学参数无明显异常,术前确诊较困难。
笔者通过复习文献,总结以下几点影像学特点:CT表现为边界清晰、密度均匀的肿块,病灶可同时累及多个部位,并且有融合倾向大,坏死少见,增强扫描轻度或显著均匀强化,肿瘤内及周边空间增粗的血管影,邻近骨质破坏,较少出现骨质硬化边;MR表现:T1多呈等低信号,T2表现为等高信号,弥散受限,ADC值减低;另外生长在血管旁的EMP还会出现特征性“夹心饼”征象,即增强扫描病变包埋邻近血管形成的征象。
鉴别诊断:本例EMP病灶位于椎旁软组织,需与发生在软组织的相关病变鉴别。首先EMP具有类似于
文献报道增强扫描EMP病灶内部可见数量及形态各异、强化更为显著的间隔影,该影像学征象在组织学上对应为血管丰富的疏松间质结构,而这一特点被归结为本病特征性影像表现。部分病例鉴别困难,需穿刺
其次,EMP需与MM晚期出现的髓外浸润相鉴别。MM常发生在老年人群,伴
另外,促结缔组织增生性小圆细胞瘤亦常表现为临床特征不明显、无明确脏器起源的多发性软组织肿块,发病年龄较轻,好发于腹腔或盆腔间隙内,尤以膀胱后间隙为著,也可表现为腹膜弥漫增厚,CT平扫病灶密度不均,边缘分叶状,且由于肿瘤生长过快,病变内常见钙化、出血、囊变、坏死,增强扫描轻中度不均匀延迟强化,早期即可发生腹腔广泛种植播散及各部位转移。软组织的转移瘤也需要与SPB相鉴别。
总之,EMP发病率低,临床及影像表现特异性不强,术前很难明确诊断。对于具有恶性淋巴瘤类似的影像学表现且与其他常见肿瘤不完全相符的病例,应考虑到EMP的可能性。该病的最终确诊“金标准”应依靠病理学检查。
来源:刘芷若,冯博.椎旁软组织髓外浆细胞瘤1例[J].中国临床医学影像杂志,2021,32(05):379-380.
- 上一篇:颅底骨原发性骨肉瘤1例
- 下一篇:64岁男子,乳腺癌改良根治术后背痛,是什么原因呢?