作者:刘春英,刘莲花,赖华,成都市妇女儿童中心医院放射科
1.病例资料
早产儿,男,窒息复苏后10余分钟;胎龄32周,顺产,出生体重2280克,
患儿母亲29岁,父亲30岁,孕期无药物及特殊物质接触史,无家族遗传及代谢性疾病;哥哥7岁,体健(鼻梁较塌)。
入院检查:实验室查
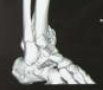
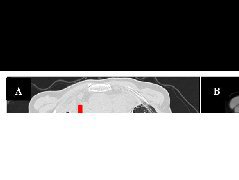
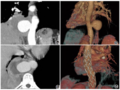
胸腹部及四肢DR:全脊柱、双侧肩胛骨、肱骨、尺骨鹰嘴(图1)、腕骨及骨盆、股骨、胫骨上端、跟骨(图2)广泛骨骺区域异常钙化灶。头颈部CT:鼻咽部狭窄,双侧

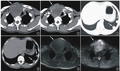
图1 DR示全脊柱、双侧肩胛骨、肱骨、尺骨鹰嘴广泛骨骺区域异常钙化灶。 图2 DR示骨盆、股骨、胫骨上端及跟骨骨骺多发钙化影。图3 CT示双侧外耳道闭锁(箭)。 图4 CT示鼻骨扁平(箭)
基因检测结果:受检者携带ARSE基因一个半合子变异,c.520C>G,p.(Pro174Ala),该变异为错义突变,使所编码蛋白质第174位氨基酸由Pro变为Ala,可能引起X连锁点状软骨发育不良。HGMD数据库未见文献报道;ESP6500siv2-ALL数据库、千人基因组(1000g2015aug-ALL)和dbSNP147数据库均未见收录;生物学软件预测其致病可能性大。患儿需继续呼吸机辅助通气治疗,家属放弃、签字出院。
2.讨论
点状软骨发育不良是一种较罕见的、在新生儿或婴儿期以骨骺软骨不规则异常钙盐沉积为病理特征的一类骨发育异常疾病。按其临床表现和遗传方式差异分5型:肢根型(常
本文报道该患儿基因结果示ARSE基因半合子变异,c.520C>G,p.(Pro174Ala),在HGMD专业版及多个数据库中均未见收录,其父母及哥哥拒绝基因检测,无法获得遗传学资料,但专家验证分析,预测其致病(CDPX1型)可能性大。遗传学检查作为点状软骨发育不良的重要确诊和分型手段,据报道在CDPX1患儿中,常规核型分析可发现25%的患儿X染色体短臂缺失或重排,在符合临床诊断标准的60%~75%患儿基因检查发现ARSE基因突变。
ARSE基因是硫酸酯酶的家族成员,其依赖
X线检查是CDPX1的重要诊断方法,骨骺正常骨化前出现异常钙化灶,如脊柱、肩胛骨、长骨、四肢关节及气管软骨。对比王彩君等总结既往报道点状发育不良病例,该患儿骨骺异常钙化灶范围最广,包括全脊柱、肩胛骨、肱骨、尺骨、股骨、胫骨并累及肘、腕、髋、膝、踝、气管软骨,出生后
目前,CDPX1无特异性治疗方案,对症治疗为主,大多数患儿预后良好,但呼吸困难、气道梗阻患儿需早期气管插管并有创机械通气,并行镜检明确气道闭锁及狭窄程度。
来源:刘春英,刘莲花,赖华.新生儿点状软骨发育不良一例[J].放射学实践,2022,37(01):137-138.